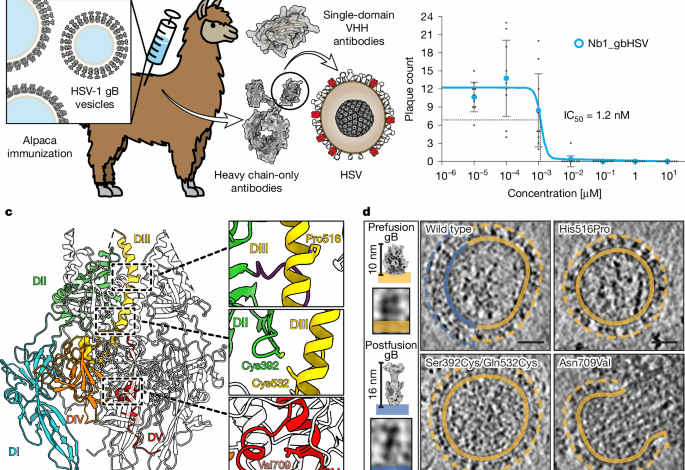

Alpaca immunization, generation of the nanobody library and phage display selection
gB-presenting vesicles were used for alpaca immunization. Generation of the nanobody library and phage display selection were performed essentially as described previously40 using biotinylated gB vesicles as baits. Recovered clones were identified by sequencing, and representatives of all classes were expressed as described below.
Cytoplasmic bacterial expression and purification of VHH antibodies
VHH antibodies were produced as His14–ScSUMO fusions by cytoplasmic expression in E. coli SHuffle Express (New England Biolabs), which allows for formation of disulfide bonds in the cytoplasm. Cells were grown in 300 ml Terrific Broth, and protein expression was induced at 21 °C with 0.08 mM isopropyl β-d-1-thiogalactopyranoside. Five hours after induction, 5 mM EDTA was added to the culture medium. The bacteria were then pelleted, resuspended in 50 mM Tris-HCl pH 7.5, 20 mM imidazole, 300 mM NaCl (lysis buffer) and frozen in liquid nitrogen. Cells were lysed by thawing and sonication, and insoluble material was removed by ultracentrifugation at around 160,000g (approximately 1 h, T647.5 rotor; Thermo Fisher Scientific).
Soluble material was applied to a 1 ml Ni2+ chelate column. The matrix was washed with lysis buffer, with further washing steps including 0.2% (w/v) Triton X‐100 or 1 M NaCl, respectively. The VHH antibody was eluted by cleaving the His14–SUMO‐tag using 100 nM Saccharomyces cerevisiae Ulp1p for 2 h at room temperature. The eluted VHH antibodies were frozen in liquid nitrogen and stored at −70 °C until further use.
Labelling of nanobodies
Nanobodies were labelled with an engineered C-terminal cysteine using maleimides of Alexa Fluor 488 or Alexa Fluor 647 for immunofluorescence as described previously40.
Mammalian cell lines
All mammalian cell lines were acquired originally from the American Type Culture Collection (ATCC). Authenticity of cell lines was confirmed through morphology and growth behaviour in cell-type-specific media. All cell lines were tested regularly for mycoplasma contamination and all results were negative. No commonly misidentified cell lines (according ICLAC and NCBI Biosample) were used in this study.
Neutralization tests and IC50 determination
To test for neutralization of the nanobody, plaque reduction assays were performed. Vero cells (ATCC, CCL-81) were seeded in 24-well plates and grown overnight to form a confluent cell layer. Before cell layer infection using around 20 plaque-forming units per well (160 plaque-forming units ml−1), viruses were pre-incubated for 30 min at room temperature with concentrations ranging from 1 nM to 1 µM of nanobodies. Subsequently, infection was performed for 1.5 h at room temperature before inoculum was removed and cells were washed with PBS. After buffer removal, cells were overlaid with 1.2% avicell in Dulbecco’s Modified Eagle Medium and 5% fetal bovine serum (FBS) to prevent secondary infection through the cell medium and to promote cell-to-cell spread (plaque formation). At 48–72 h after infection, cells were fixed with 4% paraformaldehyde (PFA) in water and stained with 0.1% crystal violet in 2% ethanol. Plaques were counted manually. IC50 values were calculated using the AAT Bioquest calculator (https://www.aatbio.com/tools/ic50-calculator).
Vesicle preparation
Vesicles were prepared as described26. In brief, BHK-21 (C13) (ATCC, CCL-10) cells were grown in Glasgow minimal essential medium (GMEM) supplemented with 20 mM HEPES pH 7.4, 2% (v/v) tryptose phosphate broth and 2% (v/v) FBS. At around 70% confluency, cells were transfected transiently with expression plasmids encoding wild-type gB (Uniprot, A1Z0P7), and derivatives thereof featuring C-terminal tags and/or mutations. Cells were grown for a further 48 h with a medium exchange to serum-free GMEM after 24 h. Vesicles were collected from the supernatant by differential centrifugation and resuspended in 20 mM HEPES pH 8, 150 mM NaCl. For alpaca immunizations, vesicles were resuspended in 20 mM HEPES pH 8, 150 mM NaCl and 250 mM sorbitol.
Expression tests
Protein expression of different double cysteine mutants was determined by testing for cell surface localization of gB. BHK-21 cells were grown in Ibidi µ-Slide eight-well chambered coverslips for 24 h before transient transfection using Lipofectamine 2000. Cells were transfected with plasmids for expression of wild-type HSV-1 gB or double cysteine mutants. HSV-1 gB (1–852), unable to localize to the surface, was used as negative control. At 24 h post transfection, cells were stained with 0.5 µM of Alexa 647 labelled Nb1_gbHSV in GMEM for 1 h at 37 °C. Next, cells were washed with PBS before staining for 10 min with 1 μg ml−1 Hoechst stain. After two PBS washes, cells were fixed with 4% PFA and imaged subsequently in PBS on a Leica DMi8 system at ×40 magnification. Surface staining efficiency was determined by counting nuclei and Alexa 647-stained cells in five random images per well. The percentage of stained cells relative to the percentage for wild-type gB was calculated for three independent experiments.
Protein expression of different gB mutants in vesicles and whole-cell extracts was tested as described in ref. 16 using SDS–PAGE and western blotting with a 1:5,000 dilution of rabbit anti-His6 antibody (Abcam) followed by a 1:5,000 dilution of anti-rabbit-HRP (Sigma-Aldrich Chemie GmbH). Twin-Strep tagged proteins were detected using a 1:4,000 dilution of Strep-Tactin HRP (iba Lifesciences). As a loading control, western blots were re-probed using a 1:2,500 dilution of mouse anti-GAPDH antibody (Sigma-Aldrich Chemie GmbH) followed by a 1:5,000 dilution of anti-mouse-HRP (Sigma-Aldrich Chemie GmbH); uncropped blots are provided in Supplementary Fig. 10.
Cross-reactivity tests
BHK-21 cells were grown in Ibidi µ-Slide eight-well chambered coverslips for 24 h before transient transfection using Lipofectamine 2000. Cells were transfected with plasmids for expression of different gB homologues, namely HSV-1, HSV-2, VZV, HCMV or EBV. In addition, as negative control, the structurally related glycoprotein G from vesicular stomatitis virus was transfected. All constructs are based on the respective wild-type sequence, and encode a C-terminally added fluorescent sfEGFP tag. An amount of 1 µg DNA + 1 µl lipofectamine per well in 200 µl GMEM was used for transfection. At 24 h post transfection, cells were washed with PBS before addition of 200 µl fresh GMEM including 0.5 µM of Alexa 647 labelled Nb1_gbHSV nanobody. After 1 h incubation at 37 °C, cells were washed with PBS and fixed subsequently in 4% PFA for 10 min. Cells were kept in PBS for imaging. Images were analysed using FIJI software48 and the JaCoP plugin49 was used to calculate Pearson’s correlation coefficients of the measured fluorescent signal in the green and red channel.
Protein purification and reconstitution
HEK293T cells (ATCC, CRL-3216) were transduced stably as described in ref. 31 with mutationally prefusion-stabilized HSV-1 gB or wild-type HSV-2 gB featuring a C-terminal Twin-Strep-tag. For co-expression, cells were previously transduced with a construct for Nb1_gbHSV featuring a signal peptide derived from HSV-1 glycoprotein H and a C-terminal His6 tag. To test for transduction efficiency, a separate reading frame on the same vector behind an internal ribosome entry site encodes for emerald green fluorescent protein. Expression vectors were obtained from Addgene (113901 and 113888). Stable cells were grown for 3–5 days in F12 medium (Gibco) with 5% FBS after reaching confluency before detachment in cold PBS. Cells were pelleted and snap-frozen in liquid nitrogen. Frozen pellets were resuspended in 20 mM HEPES, 500 mM NaCl and 50% glycerol using a douncer. A final concentration of 1.5% dodecyl maltoside and 0.15% cholesteryl hemisuccinate (Anatrace) was used for solubilization. After supernatant clearance, affinity purification was done using Strep-Tactin XT 4Flow high capacity resin (IBA). Reconstitution in peptidiscs32 (PEPTIDISC BIOTECH) was achieved by dialysis of detergent-purified protein with peptide in the presence of Bio-Bead SM-2 resin (Bio-Rad). Size exclusion chromatography (SEC) was done on a customized AEKTA Pure (Cytiva) in 20 mM HEPES, 300 mM NaCl, 5 mM arginine, 5 mM glutamate using a Superose 6 Inc. 3.2/300 column. Peak fractions were pooled and concentrated using Amicon Ultra 0.5 ml 100 kDa molecular weight cut-off spin concentrators. For nanobody co-expressed gB, solubilization and washing steps were done in the presence of 1 µM Nb1_gbHSV. The IgV domain of PILRa was expressed with an N-terminal His14-NEDD8 tag in BL21DE Rosetta2 cells using autoinduction in Terrific Broth medium overnight at 25 °C. After pelleting at 10,000g for 20 min, cells were resuspended in buffer A (50 mM HEPES pH 7.5, 500 mM NaCl, 30 mM imidazole) lysed in an LM10 Microfluidizer (Microfluidics) before spinning at 30,000g for 30 min. Supernatant was incubated with NiNTA Sepharose HP (Cytiva) for 2 h and applied to a column for washing steps with buffer A using more than 25× the bead volume. Bound protein was eluted with 2.5× the bead volume using 500 mM imidazole in buffer A. After addition of 1 µM final concentration of bdNEDP1, the mixture was dialysed overnight against buffer A. Dialysed eluate was incubated with the same volume of NiNTA Sepharose HP for 2 h before flowthrough was collected through a column and concentrated before SEC using a Superdex 200 Inc. 10/300 column in 20 mM HEPES pH 7.8 and 300 mM NaCl. Peak fractions were pooled and snap-frozen to be stored in −70 °C before use. Uncropped gels are provided in Supplementary Fig. 11.
Cryo-EM methods
Purified gB protein (3.5 µl; roughly 2.5 µM) or a 1:20 mixture of gB with Nb1_gbHSV were added to glow discharged Quantifoil R2.1 copper grids before a 3-s blotting step followed by plunging into a liquid ethane/propane mixture using a Thermo Fisher Scientific Vitrobot Mark IV. Frozen grids were imaged using a Titan Krios microscope (Thermo Fisher Scientific) operated at 300 kV and equipped with an X-FEG electron source and a K3 direct electron detector (Gatan) with a Bioquantum post-column energy filter operated in zero-loss imaging mode with a 20 eV slit width. Data were collected in fringe-free imaging aberration-free image shift mode using SerialEM50. Details for individual datasets are summarized in Extended Data Table 2.
For protein conformation analysis on vesicles, 3.5 µl gB vesicles were mixed with 0.5 µl 5 nm nanogold and were plunged as described before for cryo-ET and single-particle analysis. Tomograms were acquired, processed and reconstructed as described previously16.
Data processing and structure determination
Particle picking was performed in WARP using a custom trained template51. Particle positions were imported in CryoSPARC52 for particle extraction and further processing. All subsequent processing steps were performed in CryoSPARC. An initial model was built using ModelAngelo53 and missing residues were added in Coot54. Initial refinement was done with Isolde55. In CCP-EM Doppio (https://www.ccpem.ac.uk/), the model was fitted in the map using MolRep56 and the structure was refined using Refmac Servalcat57 and Phenix58. Where density permitted, N-acetyl-glucosamine sugars were added to glycosylation sites using ChimeraX. These glycosylated complexes were finally refined with TEMPy-ReFF v.1.2 (ref. 59). Validation was done using MolProbity v.4.5.2 (ref. 60), FDR backbone61 and FindMySequence62. Interacting residues and surfaces were determined using the ‘protein interfaces, surfaces and assemblies’ service PISA at the European Bioinformatics Institute63 (http://www.ebi.ac.uk/pdbe/prot_int/pistart.html) and ChimeraX64. Root-mean-square deviation between postfusion HSV-1 and HSV-2 gB was calculated in ChimeraX using PDB 2GUM (ref. 33). Multiple sequence alignment for epitope conservation was performed using Clustal Omega65 and conservation score was calculated as described in refs. 66,67 using SnapGene v.8.1. Figures were prepared using ChimeraX v.1.8 and Illustrator/Photoshop 2024 (Adobe). Manders’ Overlap Coefficient analysis to evaluate the goodness of model to map fit was done using TEMPy2 (ref. 68). Structure alignments were done by computing secondary structure elements using DSSP69, and plotted against a sequence alignment of HSV-1 and HSV-2 gB using the uniprot alignment tool (Clustal Omega GUI70).
Analysis of gB conformation on vesicles
Acquired videos were imported into CryoSPARC52 for patch motion correction and export for statistical analysis. Identified vesicles were sorted into three classes on the basis of their gB conformations: prefusion only, postfusion only and mixed population. Using the total amount of counted vesicles per gB construct, the percentages of each class were calculated using Excel v.16.16.27 (Microsoft).
Nanobody epitope mapping
Cryo-EM sample preparation was done as described earlier. A 2:1 molar ratio mixture of peptidisc reconstituted prefusion-stabilized and wild-type gB with a final concentration of 0.55 µM was prepared and used for all datasets. Different nanobodies or buffer were added in 50× molar excess and incubated for 5–10 min before plunge freezing. Data acquisition and processing was performed as described earlier. Details for individual datasets are summarized in Extended Data Table 2. Particles of the two conformations were separated in two-dimensional classification and further refined by heterogeneous refinement with individually generated ab initio models. Final densities, used for difference mapping, were produced by non-uniform refinement. Difference maps between the nanobody containing samples and the apo structures were produced in ChimeraX and fitted onto the respective apo structure of gB.
Affinity measurements
Binding kinetics of nanobodies with prefusion or postfusion gB were measured using grating-coupled interferometry on a Creoptix WAVEdelta instrument (Creoptix AG)71. All measurements were performed using quasi-planar polycarboxylate polymer WAVEchips with four channels (Creoptix AG) at 25 °C. Chips were coated with Strep-Tactin XT by amine coupling using a Twin-Strep-tag capturing kit (IBA Lifesciences). Prefusion-stabilized gB or postfusion gB (ectodomain of wild type) was immobilized on separate channels on the WAVEchip using a C-terminal Twin-Strep-tag to a density of 400 pg mm−2. During gB immobilization and kinetics measurements, 20 mM HEPES, pH 8.0, 300 mM NaCl was used as running buffer. Kinetics measurements were conducted using the waveRAPID mode with the tight binder protocol with 200 nM for Nb1_gbHSV and 500 nM for Nb2_gbHSV, Nb3_gbHSV and Fab SS55 (control). After each injection of nanobody or Fab, the chip was regenerated using 3 M GuHCl (IBA Lifesciences) before re-immobilization of fresh gB protein in the respective channels. For measurement of Nb4_gbHSV and PILRα (control) binding kinetics, the waveRAPID intermediate binder protocol was used, without regeneration or further gB immobilization. Nb4_gbHSV and PILRα were used at a concentration of 1 µM. All measurements were performed three times and evaluated using the WAVEcontrol software and double referenced against the reference channel (Strep-Tactin XT coated) and a blank. Every tight binder measurement was fitted using traditional fitting and an end crop was set to 1,000 s. For intermediate binder measurements, the end crop was kept at default.
Microscale thermophoresis
To determine the binding affinity of Nb1_gbHSV to prefusion-stabilized gB, covalently labelled Nb1_gbHSV-Alexa 647 (target) was diluted to a concentration of 400 pM in 20 mM HEPES, pH 8.0, 300 mM NaCl, 0.005% Tween20 (assay buffer). Prefusion-stabilized gB (ligand) was diluted to 20 nM in assay buffer and ligand buffer (20 mM HEPES, pH 8.0, 300 mM NaCl) was diluted 1:138 in assay buffer. Then, a 1:1 dilution series of gB with 16 dilutions was prepared in the diluted ligand buffer. Finally, the gB dilutions were mixed with equal volumes of the diluted Nb1_gbHSV-Alexa 647 to final gB concentrations ranging from 10 nM to 0.3 pM. The final concentration of Nb1_gbHSV in the assay was 200 pM. The Nb1_gbHSV-Alexa 647 gB mixture was incubated for around 24 h at 4 °C in the dark. Samples were loaded into standard Monolith capillaries and measured using the Monolith 2020 pico-RED instrument (NanoTemper) at 100% excitation and medium microscale thermophoresis power at 25 °C. The data were evaluated using the MO.Control v.2 software (NanoTemper). Outliers were excluded from the data before fitting to determine binding affinities. The measurements were repeated three times.
Animal work
One female alpaca kept at the Alpaca Facility of the Max Planck Institute for Multidisciplinary Sciences (Göttingen) was immunized using gB-presenting vesicles. The alpaca project (immunizations and blood sampling) has been approved by the animal welfare authority LAVES with the reference numbers 33.9-42502-05-13A351, 33.9-42502-05-17A220 and 33.19-42502-04-22-00210.
Statistical analyses
To calculate the IC50 of Nb1_gbHSV in Fig. 1b, response curves were fitted using the equation:
$$y={rm{M}}{rm{i}}{rm{n}}+({rm{M}}{rm{a}}{rm{x}}-{rm{M}}{rm{i}}{rm{n}})/1+{(x/{{rm{I}}{rm{C}}}_{50})}^{{rm{Hill; coefficient}}}$$
using the AAT Bioquest calculator (https://www.aatbio.com/tools/ic50-calculator).
For Extended Data Fig. 1, averages, s.d. and s.e.m. were calculated using Excel v.16.16.27(201012) (Microsoft).
For Extended Data Fig. 6, the Pearson’s correlation coefficients of the measured fluorescent signals were calculated using the JaCoP plugin49 in Fiji48.
Material availability
All unique materials used are available upon request from the authors. The use of nanobodies generated in this study might be subject to a material transfer agreement.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
نشر لأول مرة على: www.nature.com
تاريخ النشر: 2025-09-03 03:00:00
الكاتب: Benjamin Vollmer
تنويه من موقع “بتوقيت بيروت”:
تم جلب هذا المحتوى بشكل آلي من المصدر:
www.nature.com
بتاريخ: 2025-09-03 03:00:00.
الآراء والمعلومات الواردة في هذا المقال لا تعبر بالضرورة عن رأي موقع “بتوقيت بيروت”، والمسؤولية الكاملة تقع على عاتق المصدر الأصلي.
ملاحظة: قد يتم استخدام الترجمة الآلية في بعض الأحيان لتوفير هذا المحتوى.
ظهرت المقالة A nanobody specific to prefusion glycoprotein B neutralizes HSV-1 and HSV-2 أولاً على بتوقيت بيروت | اخبار لبنان والعالم لحظة بلحظة 24/24 تابعونا.